AI reads Medical Literature “variant2literature”
The importance of AI
Before the existence of medical AI systems, medical professionals and genome researchers found themselves limited by the extensive amount of time and money required to compensate professionals for their work in genomic standardization, analysis, and comparing gene variants and symptoms. To add on, full genomic analysis is difficult to achieve due to the human genome being composed of over twenty thousand individual gene sequences.
AI applies to many areas of genomic analysis, such as gene-assisted diagnosis, human genome annotation, and quantification of the level of correlation between a gene variant and diseases. There’s still a long way to go before we understand all of the over twenty thousand human genes’ purposes and mechanisms and predict how gene variants affect them, but AI can assist in establishing correlation matrices and prediction models, except in cases such as genetically transmitted diseases, drug reactions and cancerous genes, where data is significantly scarcer.
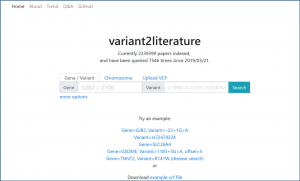
As such, we employed AI to develop “variant2literature”, which peruses a large amount of medical literature, finding related variants to diseases of interest in order to assist medical professionals in efficiently predicting possible underlying diseases correlated to the variant while raising the precision of diagnosis. In addition to finding the literature containing the variants of interest, variant2literature also provides association prediction if a variant is present along with a disease name in a single sentence.
The following paragraphs detail our methods and experimental results.
Data collection and test results
In order to determine the association between diseases and variants, initially, eatracting biomedical terms from literature is indispensable. In variant2liteature, we employed GNormPlus, tmVar and DNorm to identify the genes, variants, and diseases mentioned in PubMed Central (PMC). These tools were provided by the National Center for Biotechnology Information (NCBI), a part of the U.S. National Library of Medicine (NLM).
GNormPlus is a system that identifies gene descriptions from an excerpt. This technology is composed to two components: mention recognition and concept normalization. For mention recognition, GNormPlus uses conditional random fields (CRF) to recognize the gene descriptions. However, we still need to match the description to the gene described, which is why GNormPlus uses GenNorm in its concept normalization module to find the matching gene via vector analysis of exact matching or “bag-of-words” matching on descriptions.
tmVar is also a text-mining approach based on CRF, which is used to extract a wide range of sequence variants for both proteins and genes. These variants are defined according to a standard sequence variant nomenclature developed by the human genome variation society (HGVS). tmVar pre-processes the input text using tokenization and uses a CRF-based model to extract variant mentions for the final output.
DNorm is used to identify disease mentions, which are identified by using the BANNER named entity recognizer, a trainable system that also uses CRF. Mentions that are outputted by BANNER are then used to achieve disease normalization and identification using pairwise learning to rank.
Finally, a Recurrent Neural Network (RNN) deep learning model is used to predict the association between variants and diseases. This model was trained from a small set of literature annotated by our experts. We identified genes, variants, and diseases from these articles and categorized the types of relationships. If the instances of diseases and variants are observed in the same sentence, experts will denote whether they (a pair of variant and disease) are correlated, “Y” for yes and “O” for no. In other words, this is a machine learning algorithm that adopts binary classification. These labeled sentences will first pass through our self-trained Word2Vec, which converts the tokenized sentence into vectors, inputs these vectors into our RNN model, and then outputs the relationship between the two instances (“Y” or “O”). After training, this RNN model can be applied to the entirety of PMCOA to identify all relationships between diseases and variants.
Application
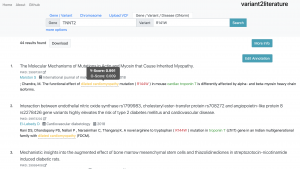
On variant2literature, the user will input either a gene or a variant. variant2literature will normalize these inputs, search for the input in the indexed papers, then output related papers and label the relevant genes, variants, and diseases in the reported papers.If a disease and a gene variant both appear within the same sentence, variant2literature will determine the correlation between the two based on the surrounding context.
Through AI-assisted analysis, variant2literature automatically determines the correlation between a disease and a gene variant, greatly reducing the time spent comparing data, reducing the cost of gene analysis, allowing medical professionals to efficiently detect underlying diseases and setting a new milstone for Taiwanese disease-related genetic testing.
